Constrained Single-Trait Phenotypic Selection#
In the Single-Trait Phenotypic Selection example, we conducted a simulation where we selected the top individuals based on their estimated breeding values. Unfortunately, this “greedy” selection strategy quickly depletes genetic diversity, leading to poorer genetic gains in the long run. One method to combat the depletion of genetic diversity is to add an inbreeding constraint. Instead of selecting the top individuals, we select the top individuals given that their mean relatedness does not exceed a specific value. In the example below, we perform both constrained and unconstrained selection and compare their long-term effects using simulations.
Simulation Preliminaries#
Loading Required Modules and Seeding the global PRNG#
To begin, we import the various modules we will be used into the Python namespace. We also set the seed for our simulation so that we can replicate the results at a later time.
# import libraries
from numbers import Real
import numpy
import pandas
import pybrops
from matplotlib import pyplot
from pybrops.breed.prot.bv.MeanPhenotypicBreedingValue import MeanPhenotypicBreedingValue
from pybrops.breed.prot.mate.TwoWayCross import TwoWayCross
from pybrops.breed.prot.mate.TwoWayDHCross import TwoWayDHCross
from pybrops.breed.prot.pt.G_E_Phenotyping import G_E_Phenotyping
from pybrops.breed.prot.sel.EstimatedBreedingValueSelection import EstimatedBreedingValueSubsetSelection
from pybrops.breed.prot.sel.OptimalContributionSelection import OptimalContributionSubsetSelection
from pybrops.model.gmod.DenseAdditiveLinearGenomicModel import DenseAdditiveLinearGenomicModel
from pybrops.opt.algo.SortingSubsetOptimizationAlgorithm import SortingSubsetOptimizationAlgorithm
from pybrops.opt.algo.SteepestDescentSubsetHillClimber import SteepestDescentSubsetHillClimber
from pybrops.popgen.bvmat.DenseBreedingValueMatrix import DenseBreedingValueMatrix
from pybrops.popgen.cmat.fcty.DenseMolecularCoancestryMatrixFactory import DenseMolecularCoancestryMatrixFactory
from pybrops.popgen.gmap.HaldaneMapFunction import HaldaneMapFunction
from pybrops.popgen.gmap.StandardGeneticMap import StandardGeneticMap
from pybrops.popgen.gmat.DensePhasedGenotypeMatrix import DensePhasedGenotypeMatrix
# seed python random and numpy random
pybrops.core.random.prng.seed(52347529)
Simulation Parameters#
Next, we define a couple of simulation parameter constants.
nfndr = 40 # number of founder individuals
nqtl = 1000 # number of QTL
qlen = 6 # length of the queue
ncross = 20 # number of cross configurations
nparent = 2 # number of parents per cross configuration
nmating = 1 # number of times to perform cross configuration
nprogeny = 80 # number of progenies per cross attempt
nrandmate = 20 # number of random intermatings
nsimul = 60 # number of simulation generations
Loading Genetic Map Data from a Text File#
Then, we load genetic map data from a CSV-like file. In this example, we are using the US NAM genetic map constructed by McMullen et al. in 2009. The code below demonstrates how these data are read into a StandardGeneticMap object using the from_csv class method.
# read genetic map
gmap = StandardGeneticMap.from_csv(
"McMullen_2009_US_NAM.gmap",
vrnt_chrgrp_col = "chr",
vrnt_phypos_col = "pos",
vrnt_genpos_col = "cM",
vrnt_genpos_units = "cM",
auto_group = True,
auto_build_spline = True,
sep = "\t",
header = 0,
)
Creating a Genetic Map Function#
After loading our genetic map data, we want to create a genetic map function object which will be used to calculate recombination probabilities for our simulations. We create a simple Haldane genetic map function using the HaldaneMapFunction class.
# use Haldane map function to calculate crossover probabilities
gmapfn = HaldaneMapFunction()
Loading Genome Data from a VCF File#
Next, we load phased genetic markers from a VCF file. In this example, we are using a subset of genetic markers from the Wisconsin Maize Diversity Panel, which is composed of 942 individuals. 2000 SNPs with a minor allele frequency greater than 0.2 have been randomly selected to keep the dataset small. From the original 942 individuals and 2000 SNPs, we randomly select 40 founders and 1000 SNPs to create a founding synthetic population.
# read phased genetic markers from a vcf file
fndr_pgmat = DensePhasedGenotypeMatrix.from_vcf("widiv_2000SNPs.vcf.gz", auto_group_vrnt=False)
# randomly select ``nfndr`` from which to create a founding population
ix = numpy.random.choice(fndr_pgmat.ntaxa, nfndr, replace = False)
fndr_pgmat = fndr_pgmat.select_taxa(ix)
# randomly select ``nqtl`` markers from founders
ix = numpy.random.choice(fndr_pgmat.nvrnt, nqtl, replace = False)
fndr_pgmat = fndr_pgmat.select_vrnt(ix)
# sort and group variants
fndr_pgmat.group_vrnt()
After loading the genetic markers, we interpolate the genetic map positions and the sequential marker crossover probabilities using the interp_xoprob method. We interpolate using the genetic map and genetic map function we have just created.
# interpolate genetic map positions
fndr_pgmat.interp_xoprob(gmap, gmapfn)
Constructing a Single-Trait Genomic Model#
Next, we want to construct a true genomic model that will model a single trait with a strictly additive genetic architecture. We construct a DenseAdditiveLinearGenomicModel object to do this.
# model intercepts: (1,ntrait)
beta = numpy.array([[0.0]], dtype = float)
# marker effects: (nvrnt,1)
mkreffect = numpy.random.normal(
loc = 0.0,
scale = 0.01,
size = (fndr_pgmat.nvrnt,1)
)
# trait names: (ntrait,)
trait = numpy.array(["Syn1"], dtype = object)
# create an additive linear genomic model to model traits
algmod = DenseAdditiveLinearGenomicModel(
beta = beta, # model intercepts
u_misc = None, # miscellaneous random effects
u_a = mkreffect, # random marker effects
trait = trait, # trait names
model_name = "synthetic_model", # name of the model
hyperparams = None # model parameters
)
Build Founder Populations & Run Breeding Program Burn-In#
In this next series of steps, we’ll create a founder population by randomly intermating our 40 selected founders for 20 generations. Following random intermating, we’ll create 6 cohorts of doubled haploid individuals, each cohort representing progenies produced in a single year. In this simulation, the oldest 3 cohorts will be aggregated into a "main" population to serve as selection candidates. The youngest 3 cohorts will be “in the pipeline” and will represent doubled haploid progenies which are being made. From the "main" population, the top 5% of individuals from each family will be selected to form a "cand" population. It is from this "cand" population that parents will be selected. The burn-in segment of the breeding program continues until the mean expected heterozygosity decreases to just under 0.3. After this point is reached, the simulated populations are ready for use in the main simulation.
Randomly Intermate for nrandmate Generations#
To start our random mating scheme, we first randomly pair up our 40 founders and mate them to create hybrids. Each mating event creates 80 progenies. This creates a population of size 1600.
# create 2-way cross object
mate2way = TwoWayCross()
# randomly select and pair founders
xconfig = numpy.random.choice(nfndr, nfndr, replace = False)
xconfig = xconfig.reshape(nfndr // 2, 2)
# randomly intermate ``nfndr`` founders to create initial hybrids
fndr_pgmat = mate2way.mate(
pgmat = fndr_pgmat,
xconfig = xconfig,
nmating = nmating,
nprogeny = nprogeny,
)
Then, we take our hybrid progenies, randomly pair them up with each other, and mate them. Each mating event produces 1 progeny. We randomly intermate progenies for 20 generations to create a founding population.
# randomly intermate for ``nrandmate`` generations
# each individual in the population is randomly mated with another individual
# and creates a single progeny so that the population size is held constant
for gen in range(1,nrandmate+1):
# randomly select and pair ``ntaxa`` parents
ntaxa = fndr_pgmat.ntaxa
xconfig = numpy.empty((ntaxa,2), dtype = int)
xconfig[:,0] = numpy.random.choice(ntaxa, ntaxa, replace = False)
xconfig[:,1] = numpy.random.choice(ntaxa, ntaxa, replace = False)
# randomly intermate ``ntaxa`` parents
fndr_pgmat = mate2way.mate(
pgmat = fndr_pgmat,
xconfig = xconfig,
nmating = 1,
nprogeny = 1,
)
print("Random Intermating:", gen)
Create Breeding Protocols for Burn-In#
Next, we make a 2-way DH cross protocol for our burn-in stage.
# create a 2-way DH cross object, use the counters from the 2-way cross object
mate2waydh = TwoWayDHCross(
progeny_counter = mate2way.progeny_counter,
family_counter = mate2way.family_counter,
)
We also create a genotyping protocol that converts phased genotypes to unphased genotypes.
# create a genotyping protocol
gtprot = DenseUnphasedGenotyping()
To simulate phenotypes, we create a simple \(G+E\) phenotyping protocol that phenotypes individuals in 4 locations, 1 replication each location. We use the founding population we have just created with random mating to set the narrow sense heritability at the single plot level to 0.4. With 4 replications, this increases the heritability to approximately 0.7 to begin.
# create a phenotyping protocol
ptprot = G_E_Phenotyping(algmod, 4, 1)
ptprot.set_h2(0.4, fndr_pgmat)
Next, we create a breeding value estimation protocol that simply uses means to estimate breeding value.
# create a breeding value estimation protocol
bvprot = MeanPhenotypicBreedingValue("taxa", "taxa_grp", trait)
Create a Within-Family Selection Function#
Next, we create a custom function to manually select individuals within families based on their breeding values. This function selects the top nindiv individuals within each family and outputs a set of indices corresponding to these individuals.
# define function to do within family selection based on yield
def within_family_selection(bvmat: DenseBreedingValueMatrix, nindiv: int) -> numpy.ndarray:
order = numpy.arange(bvmat.ntaxa)
value = bvmat.mat[:,0] # get yield breeding values
indices = []
groups = numpy.unique(bvmat.taxa_grp)
for group in groups:
mask = bvmat.taxa_grp == group
tmp_order = order[mask]
tmp_value = value[mask]
value_argsort = tmp_value.argsort()
ix = value_argsort[::-1][:nindiv]
indices.append(tmp_order[ix])
indices = numpy.concatenate(indices)
return indices
Create Cohort Structure#
In the next step, we create a cohort structure as described at the beginning of this section. We utilize dictionaries to keep track of our cohorts and populations.
# create a dictionary to store founder individuals
fndr_genome = {"cand":None, "main":None, "queue":[]}
fndr_geno = {"cand":None, "main":None, "queue":[]}
fndr_pheno = {"cand":None, "main":None}
fndr_bval = {"cand":None, "main":None}
fndr_gmod = {"cand":algmod, "main":algmod, "true":algmod}
In the code block below, we define a helper function to help us create initial cohorts of individuals from our starting randomly mated population.
# define a helper function to help make cohorts of individuals
def cohort(
mate2waydh: MatingProtocol,
pgmat: DensePhasedGenotypeMatrix,
ncross: int,
nparent: int,
nmating: int,
nprogeny: int
) -> DensePhasedGenotypeMatrix:
# sample indicies of individuals and reshape for input into mating protocol
xconfix = numpy.random.choice(pgmat.ntaxa, ncross * nparent, replace = False)
xconfig = xconfix.reshape(ncross, nparent)
# mate individuals
out = mate2waydh.mate(pgmat, xconfig, nmating, nprogeny)
return out
Next, we fill our cohort queue and construct the "main" and "cand" populations from the queue.
# fill queue with cohort genomes derived from randomly mating the founders
fndr_genome["queue"] = [cohort(mate2waydh, fndr_pgmat, ncross, nparent, nmating, nprogeny) for _ in range(qlen)]
# construct the main population genomes from the first three cohorts in the queue
fndr_genome["main"] = DensePhasedGenotypeMatrix.concat_taxa(fndr_genome["queue"][0:3])
# genotype individuals to fill the genotyping queue
fndr_geno["queue"] = [gtprot.genotype(genome) for genome in fndr_genome["queue"]]
# construct the main population genotypes from the first three cohorts in the queue
fndr_geno["main"] = DenseGenotypeMatrix.concat_taxa(fndr_geno["queue"][0:3])
# phenotype the main population
fndr_pheno["main"] = ptprot.phenotype(fndr_genome["main"])
# calculate breeding values for the main population
fndr_bval["main"] = bvprot.estimate(fndr_pheno["main"], fndr_geno["main"])
# calculate indices for within family selection to get parental candidates
ix = within_family_selection(fndr_bval["main"], 4) # select top 5%
# select parental candidates
fndr_genome["cand"] = fndr_genome["main"].select_taxa(ix)
fndr_geno["cand"] = fndr_geno["main"].select_taxa(ix)
fndr_bval["cand"] = fndr_bval["main"].select_taxa(ix)
Create a Burn-In Selection Protocol Object#
Next, we create a selection protocol for our burn-in loop. This selects individuals with the best breeding values for our synthetic trait.
# use a hillclimber for the single-objective optimization algorithm
soalgo = SortingSubsetOptimizationAlgorithm()
# create a selection protocol that selects based on EBVs with an inbreeding constraint
burnin_selprot = EstimatedBreedingValueSubsetSelection(
ntrait = 1, # number of expected traits
unscale = True, # unscale breeding values to human-readable format
ncross = 20, # number of cross configurations
nparent = 2, # number of parents per cross configuration
nmating = 1, # number of matings per cross configuration
nprogeny = 80, # number of progeny per mating event
nobj = 1, # number of objectives == 1 == yield
soalgo = soalgo, # use sorting algorithm to solve single-objective problem
)
Running a Population Burn-in until MEH is slightly less than 0.30#
Finally, we perform selection on our populations until the mean expected heterozygosity of the "main" population reaches slightly less than 0.30. This in effect sets the starting genetic diversity for our simulations so that they are not as dependent on the set of founders that were initially randomly selected.
i = 0
while fndr_genome["main"].meh() > 0.30:
# parental selection: select parents from parental candidates
selcfg = burnin_selprot.select(
pgmat = fndr_genome["cand"],
gmat = fndr_geno["cand"],
ptdf = fndr_pheno["cand"],
bvmat = fndr_bval["cand"],
gpmod = fndr_gmod["cand"],
t_cur = 0,
t_max = 0,
)
# mate: create new genomes; discard oldest cohort; concat new main population
new_genome = mate2waydh.mate(
pgmat = selcfg.pgmat,
xconfig = selcfg.xconfig,
nmating = selcfg.nmating,
nprogeny = selcfg.nprogeny,
)
fndr_genome["queue"].append(new_genome)
discard = fndr_genome["queue"].pop(0)
fndr_genome["main"] = DensePhasedGenotypeMatrix.concat_taxa(fndr_genome["queue"][0:3])
# evaluate: genotype new genomes; discard oldest cohort; concat new main population
new_geno = gtprot.genotype(new_genome)
fndr_geno["queue"].append(new_geno)
discard = fndr_geno["queue"].pop(0)
fndr_geno["main"] = DenseGenotypeMatrix.concat_taxa(fndr_geno["queue"][0:3])
# evaluate: phenotype main population
fndr_pheno["main"] = ptprot.phenotype(fndr_genome["main"])
# evaluate: calculate breeding values for the main population
fndr_bval["main"] = bvprot.estimate(fndr_pheno["main"], fndr_geno["main"])
# survivor selection: select parental candidate indices from main population
ix = within_family_selection(fndr_bval["main"], 4) # select top 5%
# survivor selection: select parental candidates from main population
fndr_genome["cand"] = fndr_genome["main"].select_taxa(ix)
fndr_geno["cand"] = fndr_geno["main"].select_taxa(ix)
fndr_bval["cand"] = fndr_bval["main"].select_taxa(ix)
print("Burn-in:", i+1)
i += 1
print("Starting MEH:", fndr_genome["main"].meh())
Simulation Setup#
In the sections above, we created the starting point for our simulations. In these next sections, we’ll test two selection strategies: one constrained by inbreeding, and another unconstrained by inbreeding. We’ll plot and compare the results of these simulations.
Create a Constrained Selection Protocol Object#
First, we’ll define a selection protocol where individuals are selected on their breeding values subject to an inbreeding constraint specified by the user. We’ll use the OptimalContributionSubsetSelection class to accomplish this task and provide objective and inequality constraint violation transformation functions to create such a scenario.
# create a dense molecular coancestry matrix factory
cmatfcty = DenseMolecularCoancestryMatrixFactory()
# define an objective transformation function
def obj_trans(
decnvec: numpy.ndarray,
latentvec: numpy.ndarray,
**kwargs: dict
) -> numpy.ndarray:
"""
Receive an incoming vector of [MGR,BV1,...,BVn] and transform it to
[BV1,...,BVn].
Where::
- MGR is the mean genomic relationship (kinship; in range [0,1]).
- BVn is the nth mean breeding value for the subset.
Parameters
----------
decnvec : numpy.ndarray
A decision space vector of shape (ndecn,)
latentvec : numpy.ndarray
A latent space function vector of shape (1+ntrait,)
Returns
-------
out : numpy.ndarray
A vector of shape (ntrait,).
"""
# extract trait(s) as objective(s)
return latentvec[1:]
# define an inequality constraint violation function
def ineqcv_trans(
decnvec: numpy.ndarray,
latentvec: numpy.ndarray,
maxinb: Real,
**kwargs: dict
) -> numpy.ndarray:
"""
A custom inequality constraint violation function.
Parameters
----------
decnvec : numpy.ndarray
A decision space vector of shape (ndecn,)
latentvec : numpy.ndarray
A latent space function vector of shape (1+ntrait,)
minvec : numpy.ndarray
Vector of minimum values for which the latent vector can take.
Returns
-------
out : numpy.ndarray
An inequality constraint violation vector of shape (1,).
"""
# calculate constraint violation for inbreeding
out = numpy.array([max(latentvec[0] - maxinb, 0.0)], dtype = float)
# return inequality constraint violation array
return out
# use a hillclimber for the single-objective optimization algorithm
soalgo = SteepestDescentSubsetHillClimber()
# create a selection protocol that selects based on EBVs with an inbreeding constraint
const_selprot = OptimalContributionSubsetSelection(
ntrait = 1, # number of expected traits
cmatfcty = cmatfcty, # coancestry/kinship matrix factory
unscale = True, # unscale breeding values to human-readable format
ncross = 20, # number of cross configurations
nparent = 2, # number of parents per cross configuration
nmating = 1, # number of matings per cross configuration
nprogeny = 80, # number of progeny per mating event
nobj = 1, # number of objectives == ntrait
obj_trans = obj_trans, # latent vector transformation to create objective function
nineqcv = 1, # number of inequality constraint violations
ineqcv_trans = ineqcv_trans, # latent vector transformation to create inequality constraints
ineqcv_trans_kwargs = { # keyword arguments
"maxinb": 1.0
},
soalgo = soalgo, # use hillclimber to solve single-objective problem
)
Create an Unconstrained Selection Protocol Object#
Second, we’ll define a selection protocol where individuals are selected solely on their breeding values. We’ll use the EstimatedBreedingValueSubsetSelection to accomplish this task.
# use a sorting algorithm for the single-objective optimization algorithm
soalgo = SortingSubsetOptimizationAlgorithm()
# create a selection protocol that selects based on EBVs with an inbreeding constraint
unconst_selprot = EstimatedBreedingValueSubsetSelection(
ntrait = 1, # number of expected traits
unscale = True, # unscale breeding values to human-readable format
ncross = 20, # number of cross configurations
nparent = 2, # number of parents per cross configuration
nmating = 1, # number of matings per cross configuration
nprogeny = 80, # number of progeny per mating event
nobj = 1, # number of objectives == ntrait
soalgo = soalgo, # use sorting algorithm to solve single-objective problem
)
Make a Statistics Recording Helper Function#
To assist in record keeping, we’ll create a function to help us record simulation metrics and store them into a dictionary.
# make recording helper function
def record(lbook: dict, gen: int, genome: dict, geno: dict, pheno: dict, bval: dict, gmod: dict) -> None:
lbook["gen"].append(gen)
lbook["main_meh"].append(genome["main"].meh())
################### main true lower selection limits ###################
tmp = gmod["true"].lsl(genome["main"], unscale = True)
lbook["main_Syn1_lsl"].append(tmp[0])
################### main true lower selection limits ###################
tmp = gmod["true"].usl(genome["main"], unscale = True)
lbook["main_Syn1_usl"].append(tmp[0])
###################### main true breeding values #######################
tbv = gmod["true"].gebv(genome["main"])
################## main true breeding value minimums ###################
tmp = tbv.tmin(unscale = True)
lbook["main_Syn1_tbv_min"].append(tmp[0])
#################### main true breeding value means ####################
tmp = tbv.tmean(unscale = True)
lbook["main_Syn1_tbv_mean"].append(tmp[0])
################## main true breeding value maximums ###################
tmp = tbv.tmax(unscale = True)
lbook["main_Syn1_tbv_max"].append(tmp[0])
############# main true breeding value standard deviations #############
tmp = tbv.tstd(unscale = True)
lbook["main_Syn1_tbv_std"].append(tmp[0])
##################### main true genetic variances ######################
tmp = gmod["true"].var_A(genome["main"])
lbook["main_Syn1_tbv_var_A"].append(tmp[0])
###################### main true genic variances #######################
tmp = gmod["true"].var_a(genome["main"])
lbook["main_Syn1_tbv_var_a"].append(tmp[0])
#################### main estimated breeding values ####################
ebv = bval["main"]
################ main estimated breeding value minimums ################
tmp = ebv.tmin(unscale = True)
lbook["main_Syn1_ebv_min"].append(tmp[0])
#################### main true breeding value means ####################
tmp = ebv.tmean(unscale = True)
lbook["main_Syn1_ebv_mean"].append(tmp[0])
################## main true breeding value maximums ###################
tmp = ebv.tmax(unscale = True)
lbook["main_Syn1_ebv_max"].append(tmp[0])
############# main true breeding value standard deviations #############
tmp = ebv.tstd(unscale = True)
lbook["main_Syn1_ebv_std"].append(tmp[0])
Simulate Constrained Phenotypic Selection for 60 Generations#
The next subsections detail simulations in the constrained scenario.
Copy Founders#
First we’ll copy our founder populations so that we don’t modify them and can use them for repeated simulations.
# deep copy founder populations, bvals, etc. so we can replicate if needed
simul_genome = copy.deepcopy(fndr_genome)
simul_geno = copy.deepcopy(fndr_geno)
simul_pheno = copy.deepcopy(fndr_pheno)
simul_bval = copy.deepcopy(fndr_bval)
simul_gmod = copy.deepcopy(fndr_gmod)
Rudimentary Logbooks#
Then, we’ll make a dictionary logbook to store simulation metrics.
# make a dictionary logbook
const_lbook = {
"gen" : [],
"main_meh" : [],
"main_Syn1_lsl" : [],
"main_Syn1_usl" : [],
"main_Syn1_tbv_min" : [],
"main_Syn1_tbv_mean" : [],
"main_Syn1_tbv_max" : [],
"main_Syn1_tbv_std" : [],
"main_Syn1_tbv_var_A" : [],
"main_Syn1_tbv_var_a" : [],
"main_Syn1_ebv_min" : [],
"main_Syn1_ebv_mean" : [],
"main_Syn1_ebv_max" : [],
"main_Syn1_ebv_std" : [],
}
Simulation Main Loop#
Next, we’ll simulate our breeding program for 60 generations. We’ll start at an inbreeding constraint of 0.84 or less to begin and gradually increase the maximum allowed inbreeding to 1.0 (complete homozygosity; zero genetic diversity) by the end of the simulation.
# record initial statistics
record(const_lbook, 0, simul_genome, simul_geno, simul_pheno, simul_bval, simul_gmod)
# create constraint change over time
# 0.84 manually determined from Pareto frontier examination
maxinb = numpy.linspace(0.84, 1, nsimul+1)
# main simulation loop
for gen in range(1,nsimul+1):
# set the inbreeding constraint
const_selprot.ineqcv_trans_kwargs["maxinb"] = maxinb[gen]
# parental selection: select parents from parental candidates
selcfg = const_selprot.select(
pgmat = simul_genome["cand"],
gmat = simul_geno["cand"],
ptdf = simul_pheno["cand"],
bvmat = simul_bval["cand"],
gpmod = simul_gmod["cand"],
t_cur = 0,
t_max = 0,
)
# mate: create new genomes; discard oldest cohort; concat new main population
new_genome = mate2waydh.mate(
pgmat = selcfg.pgmat,
xconfig = selcfg.xconfig,
nmating = selcfg.nmating,
nprogeny = selcfg.nprogeny,
)
simul_genome["queue"].append(new_genome)
discard = simul_genome["queue"].pop(0)
simul_genome["main"] = DensePhasedGenotypeMatrix.concat_taxa(simul_genome["queue"][0:3])
# evaluate: genotype new genomes; discard oldest cohort; concat new main population
new_geno = gtprot.genotype(new_genome)
simul_geno["queue"].append(new_geno)
discard = simul_geno["queue"].pop(0)
simul_geno["main"] = DenseGenotypeMatrix.concat_taxa(simul_geno["queue"][0:3])
# evaluate: phenotype main population
simul_pheno["main"] = ptprot.phenotype(simul_genome["main"])
# evaluate: calculate breeding values for the main population
simul_bval["main"] = bvprot.estimate(simul_pheno["main"], simul_geno["main"])
# survivor selection: select parental candidate indices from main population
ix = within_family_selection(simul_bval["main"], 4) # select top 5%
# survivor selection: select parental candidates from main population
simul_genome["cand"] = simul_genome["main"].select_taxa(ix)
simul_geno["cand"] = simul_geno["main"].select_taxa(ix)
simul_bval["cand"] = simul_bval["main"].select_taxa(ix)
# record statistics
record(const_lbook, gen, simul_genome, simul_geno, simul_pheno, simul_bval, simul_gmod)
print("Generation:", gen)
Saving Results to a File#
Finally, we’ll save the results of our simulation to a CSV file.
# create output dataframe and save
const_lbook_df = pandas.DataFrame(const_lbook)
const_lbook_df.to_csv("const_lbook.csv", sep = ",", index = False)
Simulate Unconstrained Phenotypic Selection for 60 Generations#
The next subsections detail simulations in the unconstrained scenario.
Copy Founders#
As before, we’ll copy our founder populations so that we don’t modify them and can use them for repeated simulations.
# deep copy founder populations, bvals, etc. so we can replicate if needed
simul_genome = copy.deepcopy(fndr_genome)
simul_geno = copy.deepcopy(fndr_geno)
simul_pheno = copy.deepcopy(fndr_pheno)
simul_bval = copy.deepcopy(fndr_bval)
simul_gmod = copy.deepcopy(fndr_gmod)
Rudimentary Logbooks#
Then, we’ll make a dictionary logbook to store simulation metrics.
# make a dictionary logbook
unconst_lbook = {
"gen" : [],
"main_meh" : [],
"main_Syn1_lsl" : [],
"main_Syn1_usl" : [],
"main_Syn1_tbv_min" : [],
"main_Syn1_tbv_mean" : [],
"main_Syn1_tbv_max" : [],
"main_Syn1_tbv_std" : [],
"main_Syn1_tbv_var_A" : [],
"main_Syn1_tbv_var_a" : [],
"main_Syn1_ebv_min" : [],
"main_Syn1_ebv_mean" : [],
"main_Syn1_ebv_max" : [],
"main_Syn1_ebv_std" : [],
}
Simulation Main Loop#
Next, we’ll simulate our breeding program for 60 generations.
# record initial statistics
record(unconst_lbook, 0, simul_genome, simul_geno, simul_pheno, simul_bval, simul_gmod)
# main simulation loop
for gen in range(1,nsimul+1):
# parental selection: select parents from parental candidates
selcfg = unconst_selprot.select(
pgmat = simul_genome["cand"],
gmat = simul_geno["cand"],
ptdf = simul_pheno["cand"],
bvmat = simul_bval["cand"],
gpmod = simul_gmod["cand"],
t_cur = 0,
t_max = 0,
)
# mate: create new genomes; discard oldest cohort; concat new main population
new_genome = mate2waydh.mate(
pgmat = selcfg.pgmat,
xconfig = selcfg.xconfig,
nmating = selcfg.nmating,
nprogeny = selcfg.nprogeny,
)
simul_genome["queue"].append(new_genome)
discard = simul_genome["queue"].pop(0)
simul_genome["main"] = DensePhasedGenotypeMatrix.concat_taxa(simul_genome["queue"][0:3])
# evaluate: genotype new genomes; discard oldest cohort; concat new main population
new_geno = gtprot.genotype(new_genome)
simul_geno["queue"].append(new_geno)
discard = simul_geno["queue"].pop(0)
simul_geno["main"] = DenseGenotypeMatrix.concat_taxa(simul_geno["queue"][0:3])
# evaluate: phenotype main population
simul_pheno["main"] = ptprot.phenotype(simul_genome["main"])
# evaluate: calculate breeding values for the main population
simul_bval["main"] = bvprot.estimate(simul_pheno["main"], simul_geno["main"])
# survivor selection: select parental candidate indices from main population
ix = within_family_selection(simul_bval["main"], 4) # select top 5%
# survivor selection: select parental candidates from main population
simul_genome["cand"] = simul_genome["main"].select_taxa(ix)
simul_geno["cand"] = simul_geno["main"].select_taxa(ix)
simul_bval["cand"] = simul_bval["main"].select_taxa(ix)
# record statistics
record(unconst_lbook, gen, simul_genome, simul_geno, simul_pheno, simul_bval, simul_gmod)
print("Generation:", gen)
Saving Results to a File#
Finally, we’ll save the results of our simulation to a CSV file.
# create output dataframe and save
unconst_lbook_df = pandas.DataFrame(unconst_lbook)
unconst_lbook_df.to_csv("unconst_lbook.csv", sep = ",", index = False)
Visualizing Breeding Program Simulation Results with matplotlib#
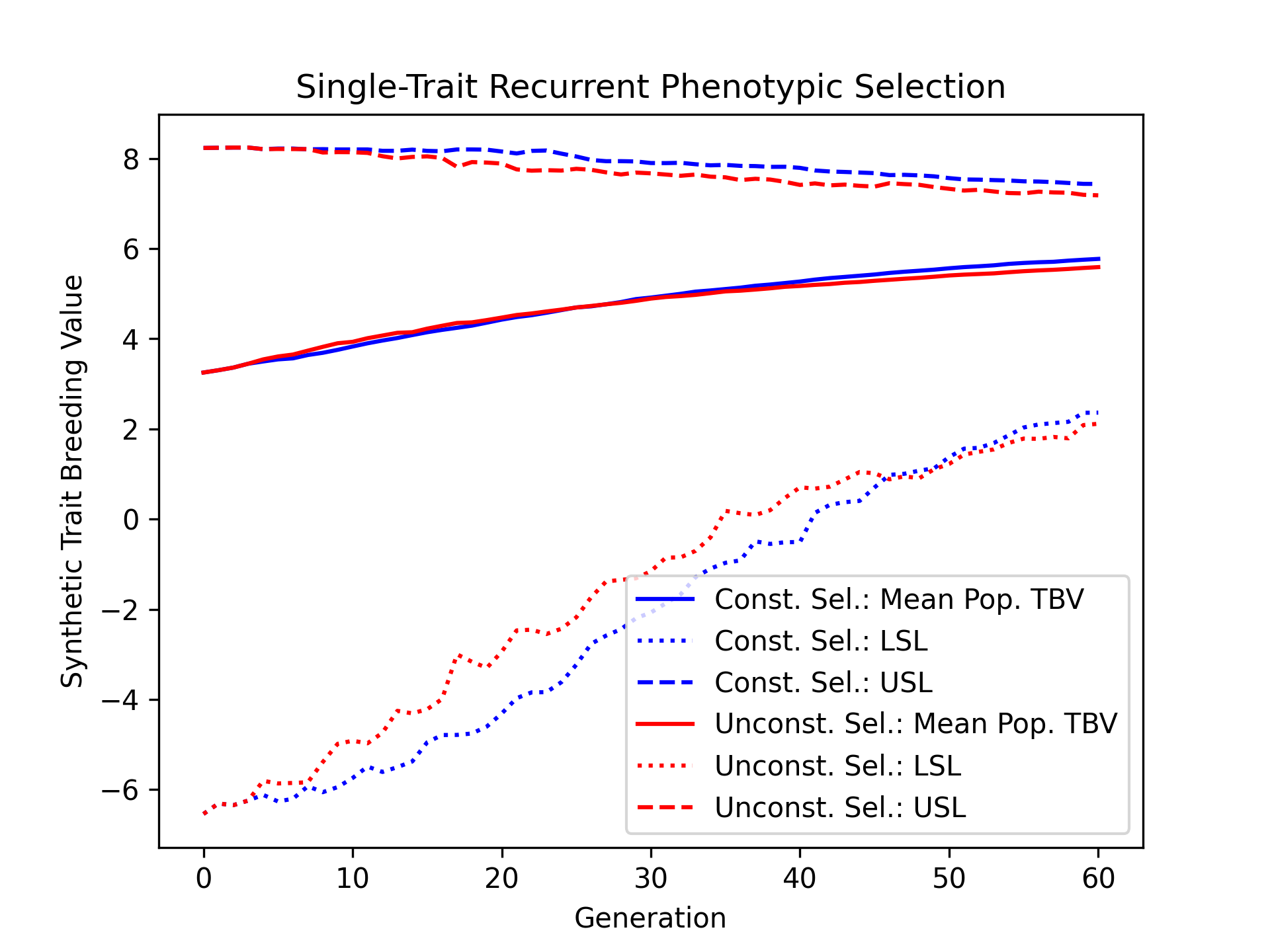
Visualizing True Breeding Values (TBVs)#
Using the results, we’ll visualize the population mean true breeding values for the constrained and unconstrained scenarios. Furthermore, we’ll plot the upper and lower selection limits to depict the narrowing of the genetic diverity over time.
# create static figure
fig = pyplot.figure()
ax = pyplot.axes()
ax.plot(const_lbook_df["gen"], const_lbook_df["main_Syn1_tbv_mean"], '-b', label = "Const. Sel.: Mean Pop. TBV")
ax.plot(const_lbook_df["gen"], const_lbook_df["main_Syn1_lsl"], ':b', label = "Const. Sel.: LSL")
ax.plot(const_lbook_df["gen"], const_lbook_df["main_Syn1_usl"], '--b', label = "Const. Sel.: USL")
ax.plot(unconst_lbook_df["gen"], unconst_lbook_df["main_Syn1_tbv_mean"], '-r', label = "Unconst. Sel.: Mean Pop. TBV")
ax.plot(unconst_lbook_df["gen"], unconst_lbook_df["main_Syn1_lsl"], ':r', label = "Unconst. Sel.: LSL")
ax.plot(unconst_lbook_df["gen"], unconst_lbook_df["main_Syn1_usl"], '--r', label = "Unconst. Sel.: USL")
ax.set_title("Single-Trait Recurrent Phenotypic Selection")
ax.set_xlabel("Generation")
ax.set_ylabel("Synthetic Trait Breeding Value")
ax.legend()
pyplot.savefig("constrained_single_trait_phenotypic_selection_true_breeding_values.png", dpi = 300)
pyplot.close(fig)
The figure below is the results of the code above.
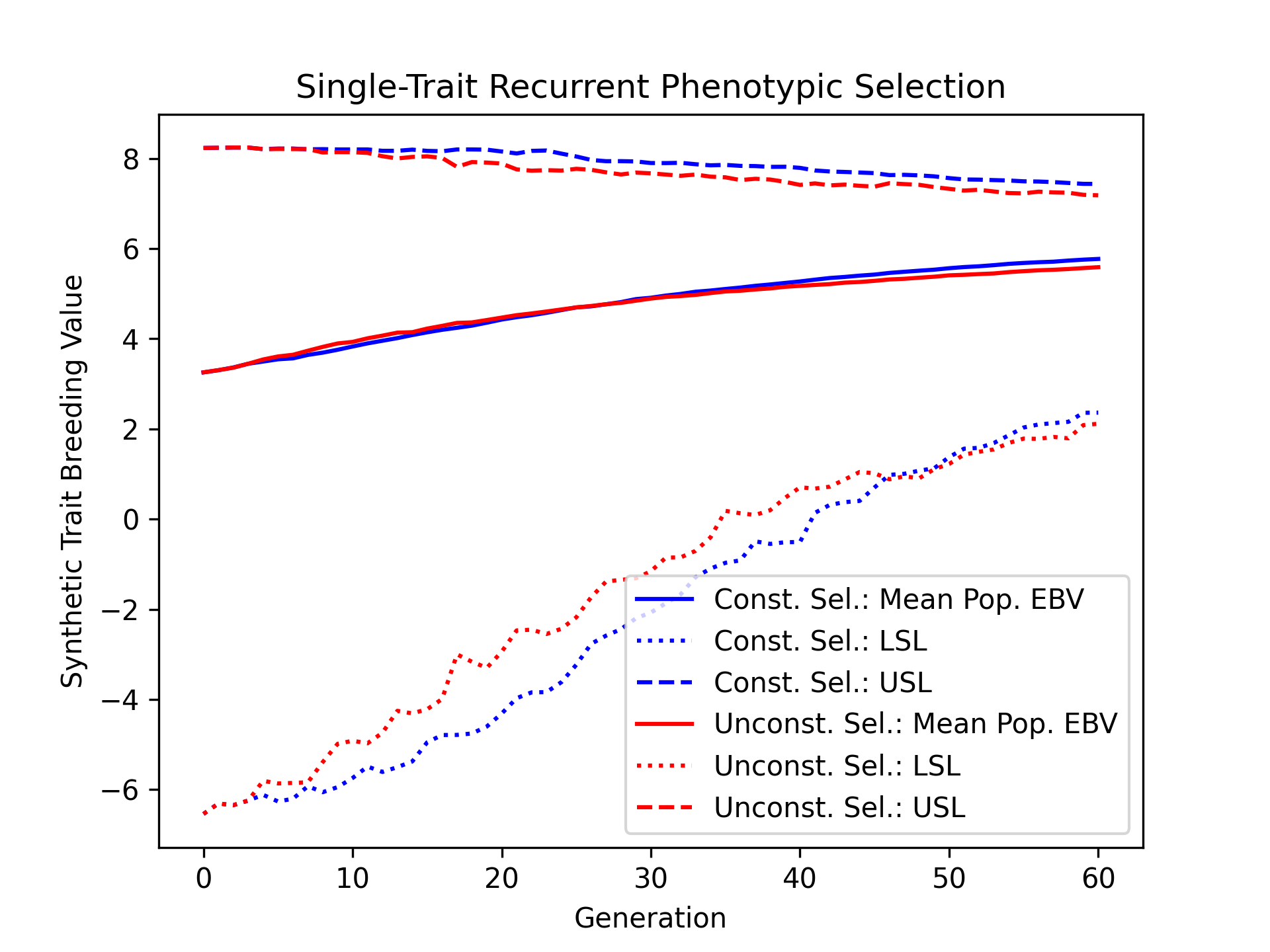
Visualizing Estimated Breeding Values (TBVs)#
Next, we’ll visualize the estimated breeding values.
# create static figure
fig = pyplot.figure()
ax = pyplot.axes()
ax.plot(const_lbook_df["gen"], const_lbook_df["main_Syn1_ebv_mean"], '-b', label = "Const. Sel.: Mean Pop. EBV")
ax.plot(const_lbook_df["gen"], const_lbook_df["main_Syn1_lsl"], ':b', label = "Const. Sel.: LSL")
ax.plot(const_lbook_df["gen"], const_lbook_df["main_Syn1_usl"], '--b', label = "Const. Sel.: USL")
ax.plot(unconst_lbook_df["gen"], unconst_lbook_df["main_Syn1_ebv_mean"], '-r', label = "Unconst. Sel.: Mean Pop. EBV")
ax.plot(unconst_lbook_df["gen"], unconst_lbook_df["main_Syn1_lsl"], ':r', label = "Unconst. Sel.: LSL")
ax.plot(unconst_lbook_df["gen"], unconst_lbook_df["main_Syn1_usl"], '--r', label = "Unconst. Sel.: USL")
ax.set_title("Single-Trait Recurrent Phenotypic Selection")
ax.set_xlabel("Generation")
ax.set_ylabel("Synthetic Trait Breeding Value")
ax.legend()
pyplot.savefig("constrained_single_trait_phenotypic_selection_estimated_breeding_values.png", dpi = 300)
pyplot.close(fig)
The figure below is the results of the code above.
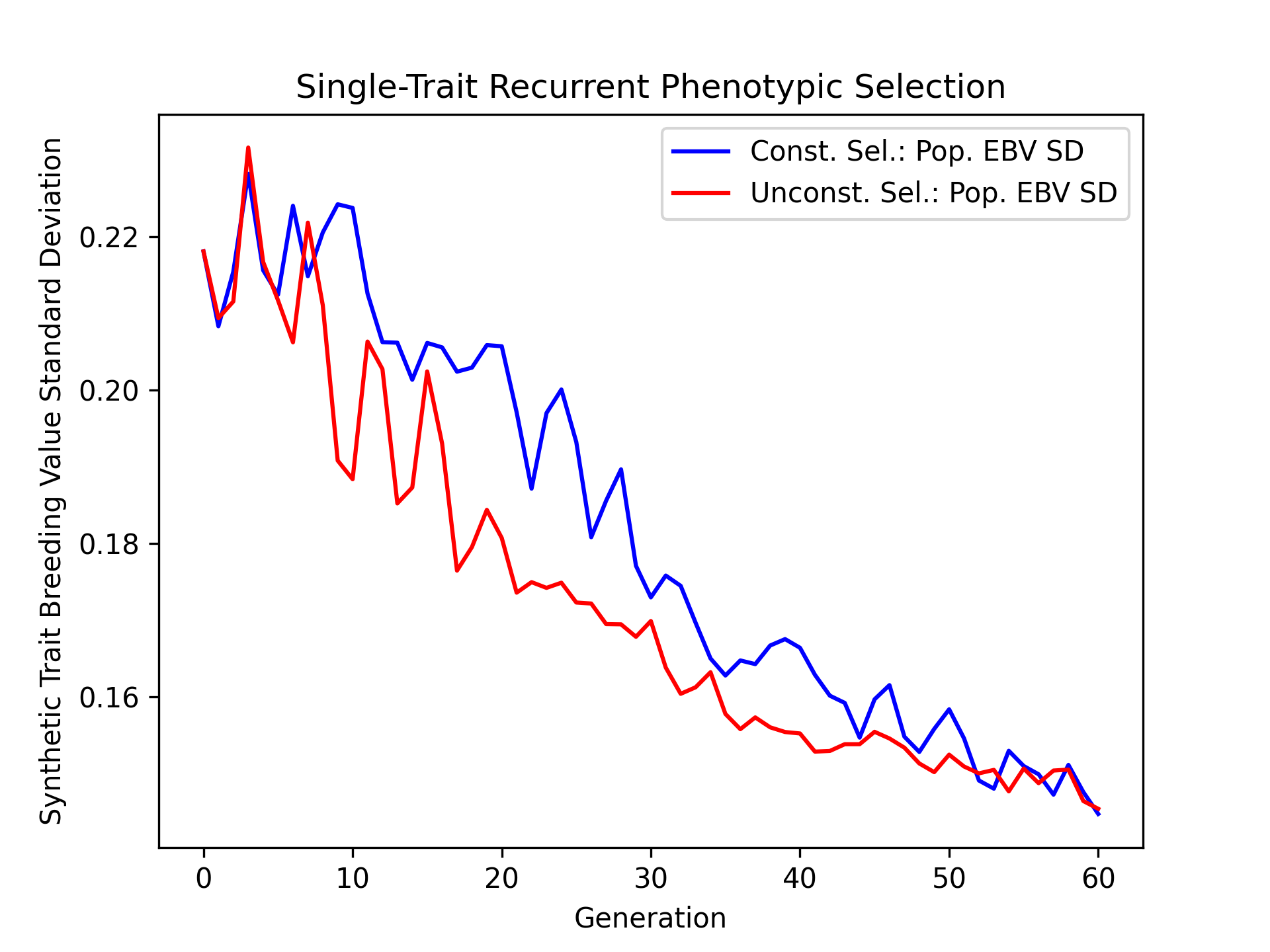
Visualizing Mean Expected Heterozygosity (MEH)#
Mean expected heterozygosity is a valuable diversity metric. We’ll plot the change in mean expected diversity over time for both the constrained and unconstrained selection scenarios.
# create static figure
fig = pyplot.figure()
ax = pyplot.axes()
ax.plot(const_lbook_df["gen"], const_lbook_df["main_meh"], '-b', label = "Const. Sel.: Pop. MEH")
ax.plot(unconst_lbook_df["gen"], unconst_lbook_df["main_meh"], '-r', label = "Unconst. Sel.: Pop. MEH")
ax.set_title("Single-Trait Recurrent Phenotypic Selection")
ax.set_xlabel("Generation")
ax.set_ylabel("Mean Expected Heterozygosity")
ax.legend()
pyplot.savefig("constrained_single_trait_phenotypic_selection_mean_expected_heterozygosity.png", dpi = 300)
pyplot.close(fig)
The figure below is the results of the code above.
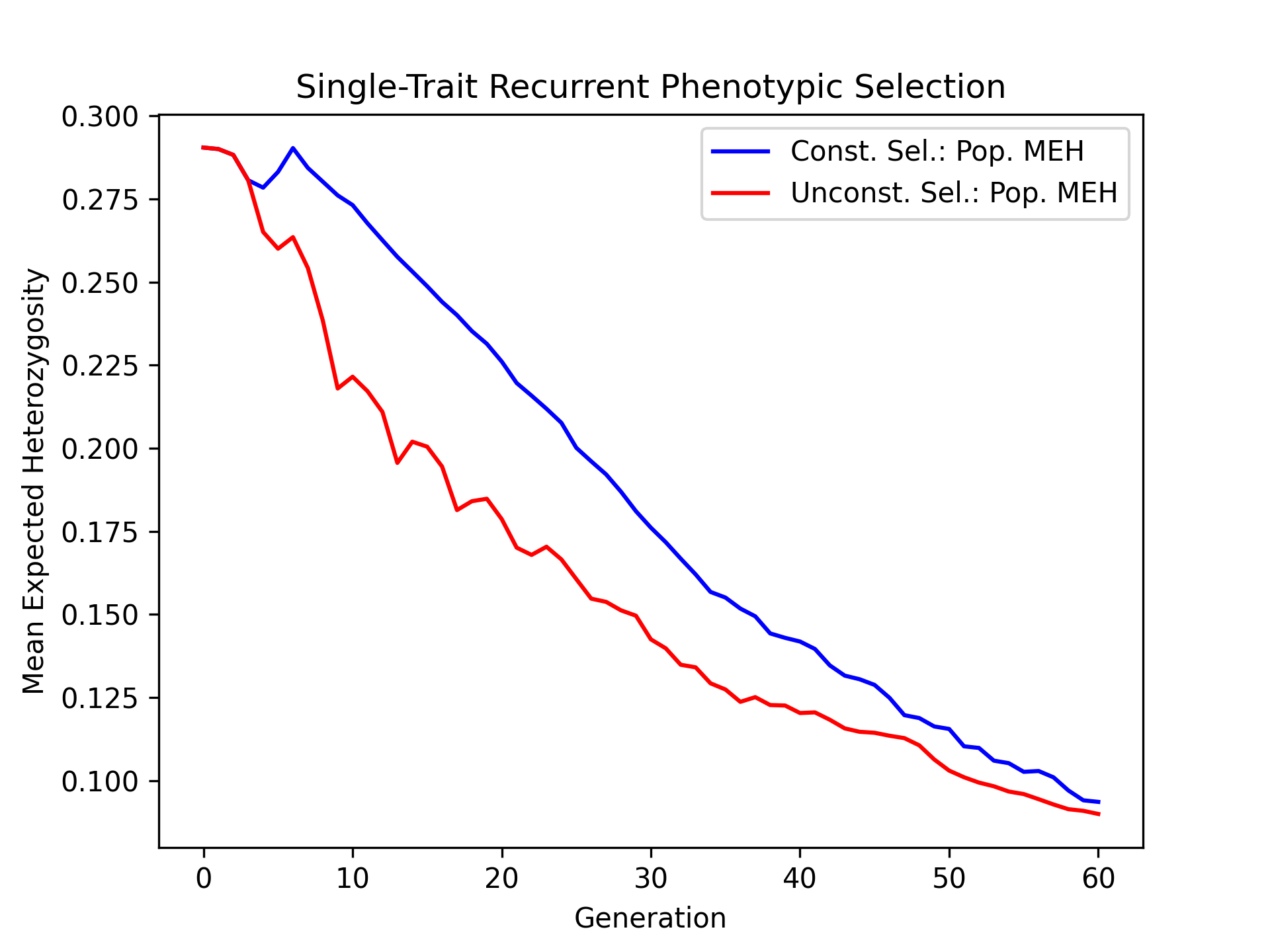
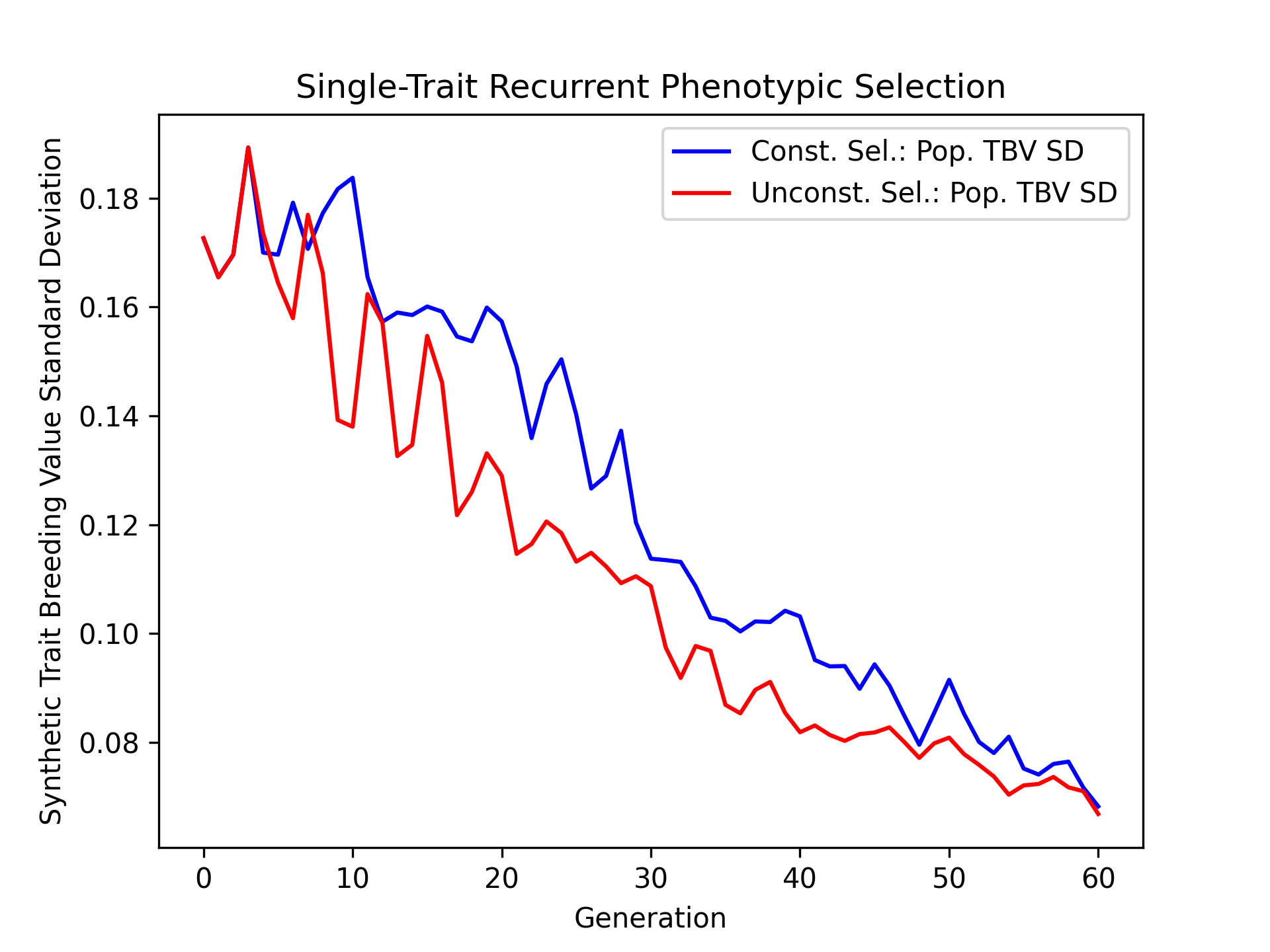
Visualizing True Breeding Value Standard Deviations#
Plotting the change in population breeding value variance is another important metric for examining the change in genetic diverity over time. We’ll plot the constrained and unconstrained population true breeding value standard deviations over time.
# create static figure
fig = pyplot.figure()
ax = pyplot.axes()
ax.plot(const_lbook_df["gen"], const_lbook_df["main_Syn1_tbv_std"], '-b', label = "Const. Sel.: Pop. TBV SD")
ax.plot(unconst_lbook_df["gen"], unconst_lbook_df["main_Syn1_tbv_std"], '-r', label = "Unconst. Sel.: Pop. TBV SD")
ax.set_title("Single-Trait Recurrent Phenotypic Selection")
ax.set_xlabel("Generation")
ax.set_ylabel("Synthetic Trait Breeding Value Standard Deviation")
ax.legend()
pyplot.savefig("constrained_single_trait_phenotypic_selection_true_breeding_value_standard_deviation.png", dpi = 300)
pyplot.close(fig)
The figure below is the results of the code above.
Visualizing Estimated Breeding Value Standard Deviations#
We’ll do the same thing as above but with estimated breeding values.
# create static figure
fig = pyplot.figure()
ax = pyplot.axes()
ax.plot(const_lbook_df["gen"], const_lbook_df["main_Syn1_ebv_std"], '-b', label = "Const. Sel.: Pop. EBV SD")
ax.plot(unconst_lbook_df["gen"], unconst_lbook_df["main_Syn1_ebv_std"], '-r', label = "Unconst. Sel.: Pop. EBV SD")
ax.set_title("Single-Trait Recurrent Phenotypic Selection")
ax.set_xlabel("Generation")
ax.set_ylabel("Synthetic Trait Breeding Value Standard Deviation")
ax.legend()
pyplot.savefig("constrained_single_trait_phenotypic_selection_estimated_breeding_value_standard_deviation.png", dpi = 300)
pyplot.close(fig)
The figure below is the results of the code above.